概要
『PIMD』は志賀基之博士(国立研究開発法人日本原子力研究開発機構・研究主幹)によって独自に開発された 並列分子シミュレーションを対象とするオープンソース・ソフトウェアです。この MPI Fortran 90 に基づくプログラムは Apache 2.0 License の範囲で、どなたでも無償でダウンロードし、ご利用になれます。
バージョン2.7.0では、N2P2とのインターフェイスが実装され、機械学習のニューラルネットワークポテンシャルを利用したシミュレーションが可能になりました。また、VASPバージョン6とのインターフェイスが実装され、第一原理密度汎関数理論を使用したシミュレーションも可能になりました。バージョン2.7.0.r2では、intel ifortの廃止に伴い makefile が変更されました。 バージョン 2.7.1 では、経路積分シミュレーションを高速化するために、並列計算のためのローカルスキーム(XMPI)が実装されました。また、汎用機械学習ポテンシャルを用いたシミュレーションを可能にするため、PFP とのインターフェースが実装されました。さらに、金属‐水界面向けの GAL21 力場も実装されました。ブラウニアン鎖分子動力学法および温度制御つきリング・ポリマー分子動力学法における乱数生成器のハードウェア依存性が修正されました。 バージョン 2.7.2 では、MACE 機械学習原子間ポテンシャルを用いたシミュレーションを可能にするため、MACE とのインターフェースが実装されました。このインターフェースは CUDA 対応の PyTorch を介した GPU 加速に対応しており、分子動力学シミュレーションおよび経路積分シミュレーションを効率的に実行するのに適しています。特徴
『PIMD』に実装された内容は以下の通りです。
シミュレーション手法
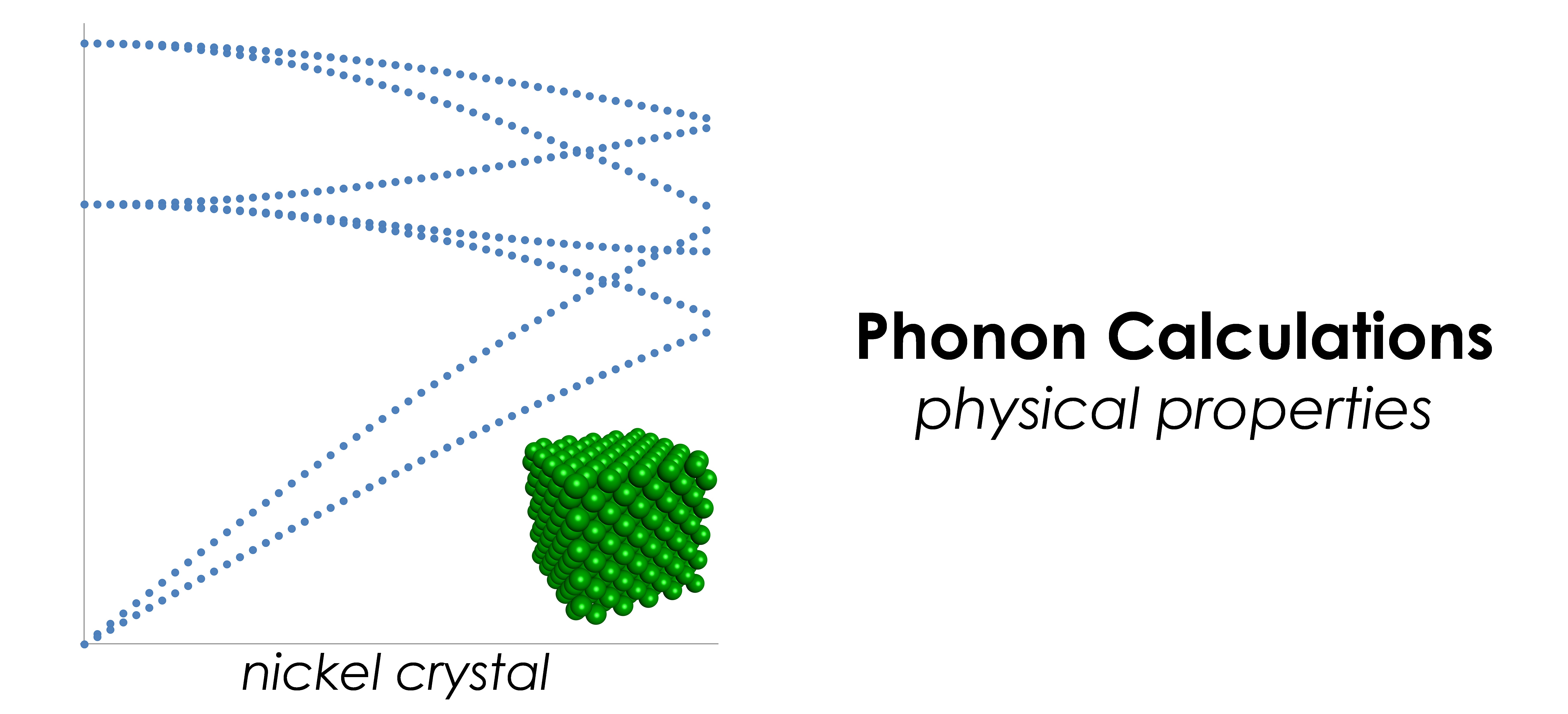
- 構造最適化、基準振動解析、フォノン計算、弾性定数
- 反応経路探索:ストリング法、最急降下法、最緩上昇法
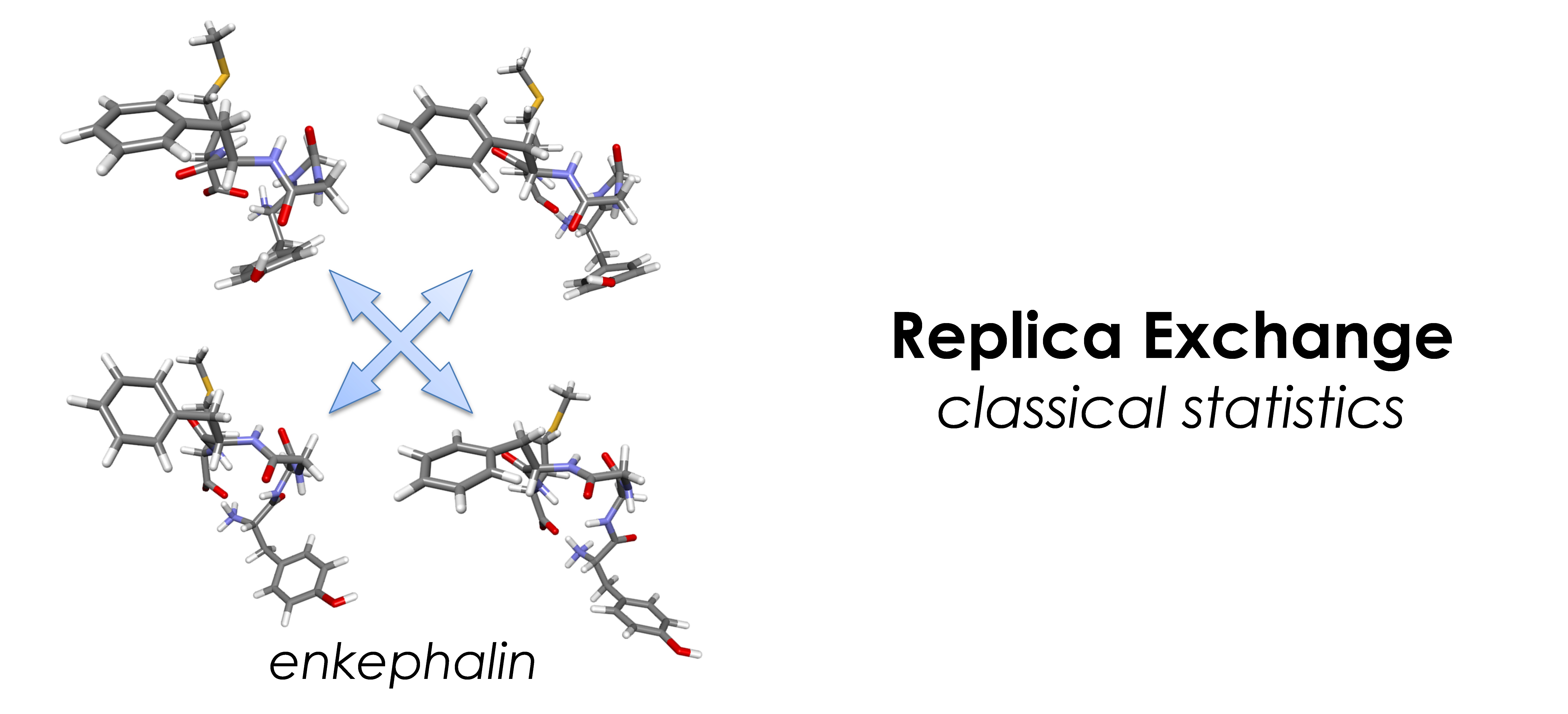
- 古典統計:レプリカ交換法
- 量子統計:経路積分分子動力学法
- 古典力学:分子動力学法、ハイブリッド・モンテカルロ法
- 半古典動力学:セントロイドおよびリング・ポリマー分子動力学法
- 半古典動力学:ブラウニアン鎖分子動力学法
- 非断熱動力学:平均場動力学法、サーフェス・ホッピング法
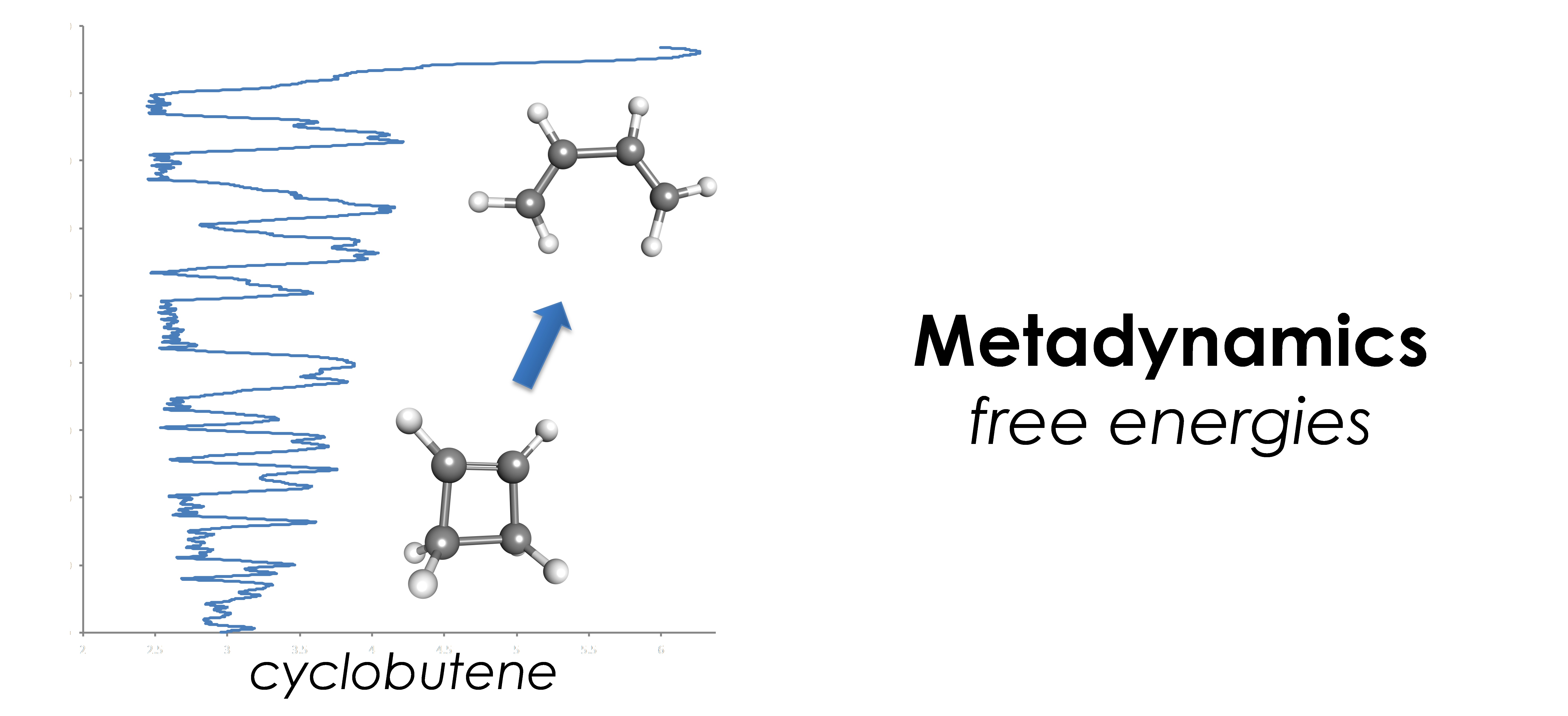
- 自由エネルギー計算:メタ・ダイナミクス法、拘束分子動力学法、平均力ダイナミクス
- 機械学習:自己学習・ハイブリッドモンテカルロ法
アンサンブル
- エネルギー一定(NVE)、温度一定(NVT)、圧力一定(NPT)、応力一定(NtT)
境界条件
- 自由境界、周期境界(立方セル、平行六面体セル)
ポテンシャル
- 第一原理的手法:SMASH
- 古典力場:ADP、AMBER、CHARMM、CLAYFF、EAM、GAL(new!)、OPLS、Tersoff
- 分極力場:誘起双極子とTholeダンピング補正
- 他のコードとのインターフェース(第一原理的手法):ABINIT-MP、CP2K、GAMESS、GAUSSIAN、MOLPRO、NTCHEM、ORCA、PHASE/0, QUANTUM ESPRESSO、TURBOMOLE、VASP
- 他のコードとのインターフェース(半経験的手法):DFTB、MOPAC、XTB
- マルチスケール法:ONIOM、QM/MM
- 他のコードとのインターフェース(機械学習ポテンシャル):AENET, MTP, N2P2, Matlantis PFP*(new!)
- ユーザー定義のサブルーチン
PIMD コードの引用文献
- M. Shiga, PIMD version 2.7.1 (2026).
- M. Shiga, M. Tachikawa, S. Miura, J. Chem. Phys. 115, 9149-9159 (2001).
"A unified scheme for ab initio molecular orbital theory and path integral molecular dynamics'' - M. Shiga, M. Tachikawa, S. Miura, Chem. Phys. Lett. 332, 396-402 (2000).
"Ab initio molecular orbital calculation considering the quantum mechanical effect of nuclei by path integral molecular dynamics''
*Matlantis PFP は開発段階のため、予告なしに対応が取り下げられる可能性があります。