About
PIMD is an open-source software for parallel molecular simulations, originally developed by Dr. Motoyuki Shiga, Principal Researcher at the Japan Atomic Energy Agency. This program, which is based on MPI Fortran 90, can be downloaded and used by anyone free of charge under the Apache 2.0 License.
In version 2.7.0, an interface with N2P2 has been implemented, enabling simulations using machine learning neural network potentials. An interface with VASP version 6 has also been implemented, enabling simulations using ab initio density functional theory. In version 2.7.0.r2, the makefile has changed due to the discontinuation of intel ifort. In version 2.7.1, a local scheme for parallel computation (XMPI) has been implemented to accelerate path integral simulations. An interface with PFP has been implemented to enable simulations using universal machine learning potentials. GAL21 force field for metal-water interface has been implemented as well. Hardware dependence on random number generators has been fixed for Brownian chain molecular dynamics and thermostatted ring polymer molecular dynamics simulations. In version 2.7.2, an interface with MACE has been implemented to enable simulations using MACE machine-learning interatomic potentials. The interface supports GPU acceleration through CUDA-enabled PyTorch, making it suitable for efficient molecular dynamics and path-integral simulations.Features
The main contents of PIMD is as follows.
Simulation methods
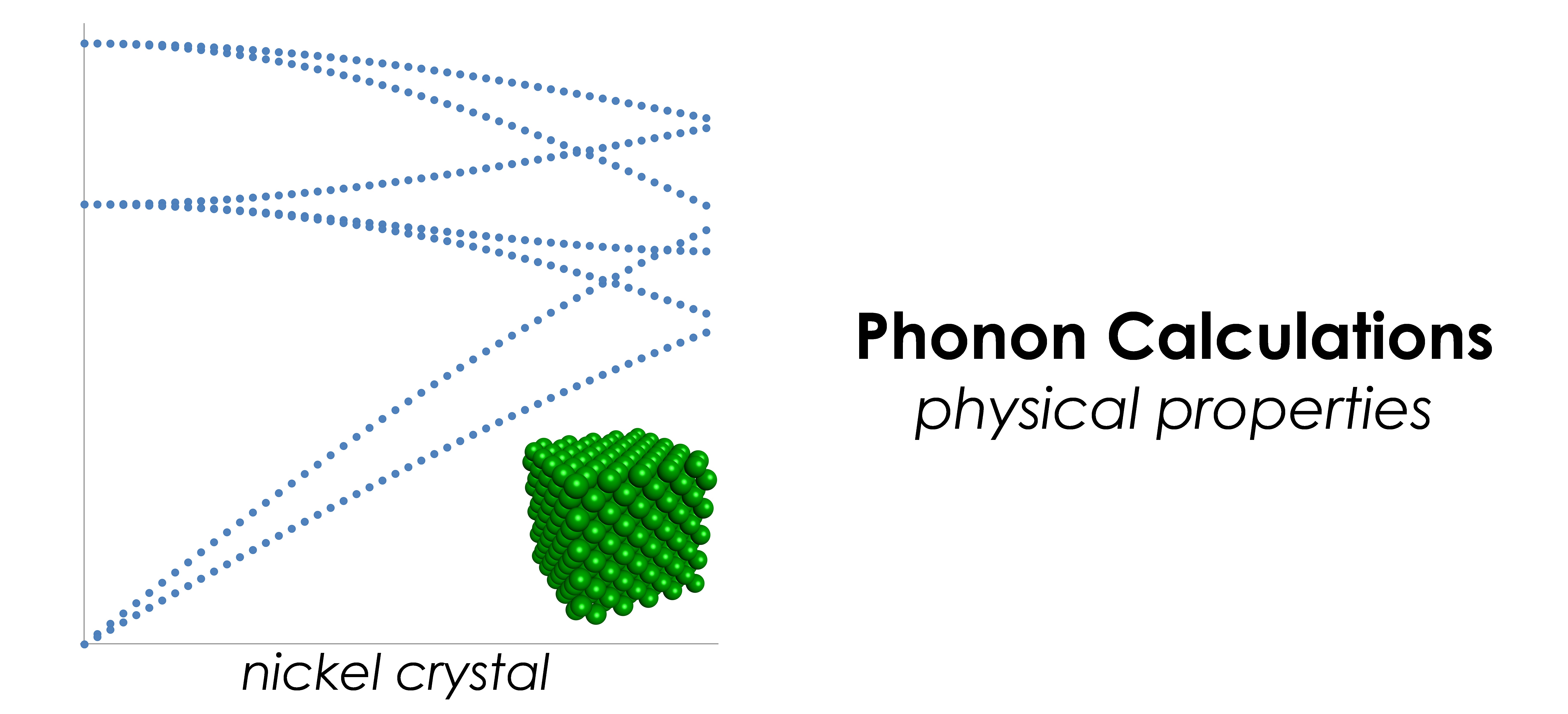
- Geometry optimization, normal mode analysis, phonon calculations, elastic constants
- Reaction paths: string method, steepest descent method, gentlest ascent method
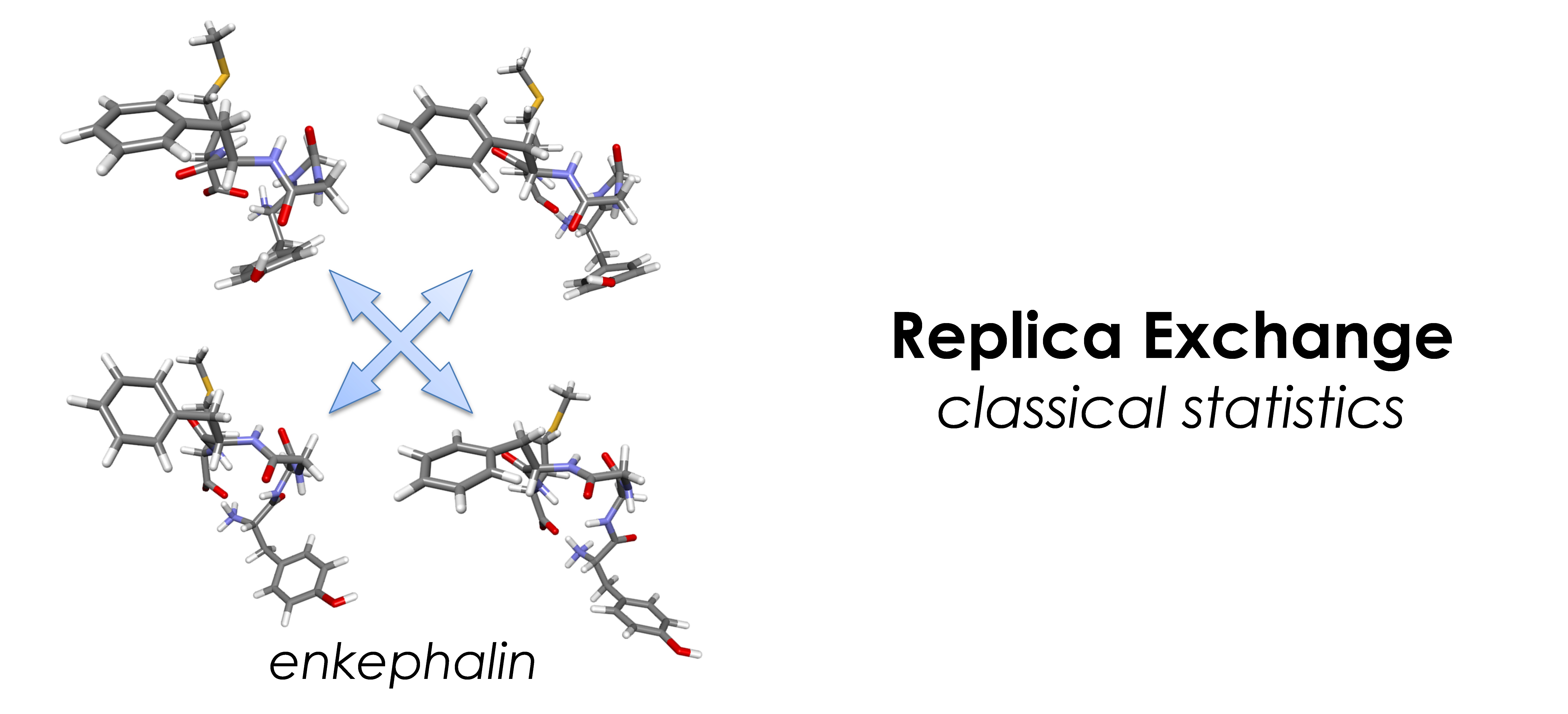
- Classical statistics: replica exchange method
- Quantum statistics: path integral molecular dynamics
- Classical mechanics: molecular dynamics, hybrid Monte Carlo
- Semiclassical dynamics: centroid and ring polymer molecular dynamics
- Nonadiabatic dynamics: mean field dynamics, surface hopping
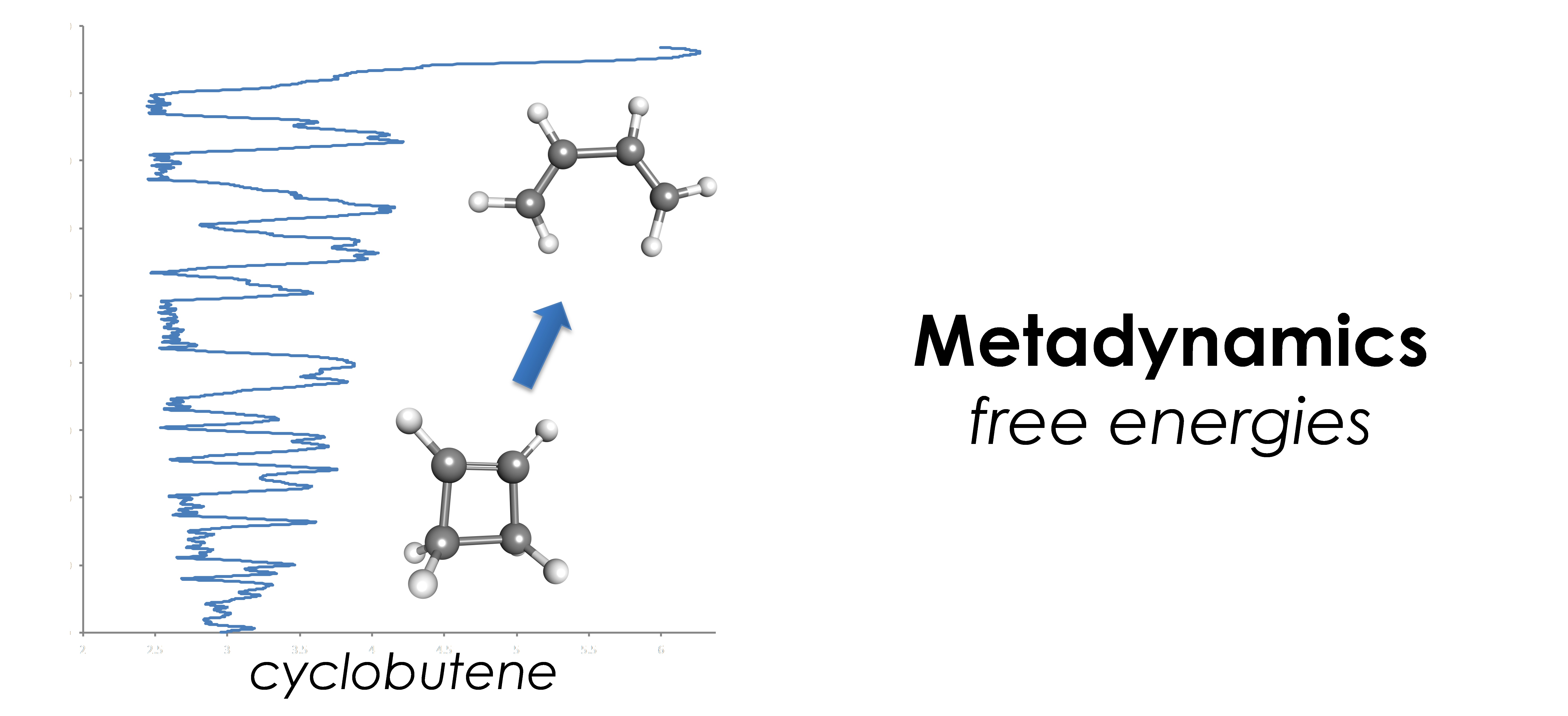
- Free energies: metadyamics, constrained molecular dynamics, mean force dynamics
- Machine Learning: Self-learning hybrid Monte Carlo
Statistical ensembles
- Constant energy (NVE), constant temperature (NVT), constant pressure (NPT), constant stress (NtT)
Boundary conditions
- Free boundary, periodic boundary (cubic cell, parallel-piped cell)
Potentials
- Ab initio quantum chemistry: SMASH
- Classical force fields: ADP, AMBER, CHARMM, CLAYFF, EAM, GAL(new!), OPLS, Tersoff
- Polarizable force field: Induced dipoles with Thole damping correction
- Interface with other codes (ab initio): ABINIT-MP, CP2K, GAMESS, GAUSSIAN, MOLPRO, ORCA, PHASE/0, QUANTUM ESPRESSO, TURBOMOLE, VASP
- Interface with other codes (semiempirical): DFTB, MOPAC, XTB
- Multiscale methods: ONIOM, QM/MM
- Interface with other codes (Machine learning potentials): AENET, MTP, N2P2, Matlantis PFP*(new!)
- A subroutine intended for user-defined potentials.
Literature references of the PIMD code:
- M. Shiga, PIMD version 2.7.1 (2026).
- M. Shiga, M. Tachikawa, S. Miura, J. Chem. Phys. 115, 9149-9159 (2001).
"A unified scheme for ab initio molecular orbital theory and path integral molecular dynamics'' - M. Shiga, M. Tachikawa, S. Miura, Chem. Phys. Lett. 332, 396-402 (2000).
"Ab initio molecular orbital calculation considering the quantum mechanical effect of nuclei by path integral molecular dynamics''
*Since Matlantis PFP is still under development, support may be withdrawn without prior notice.